PRÁCTICA FAMILIAR RURAL
│Vol.5│No.3│Noviembre 2020
Cómo citar este artículo
Miranda, K., Ochoa, M., Giler, N. Bonifaz, M. Lipodistrofia Congénita Generalizada. Informe de caso. Práctica Familiar Rural. 2020 noviembre; 5(3).
NÚMEROS ANTERIORES
CASOS CLÍNICOS Y EJERCICIOS CLÍNICO PATOLÓGICOS
Lipodistrofia Congénita Generalizada.
Informe de caso
Kevin Miranda Suárez[1], Miguel J. Ochoa Andrade[2}, Natasha Giler[3], Marco Bonifaz Valverde[4]
1. Novaclínica Santa Cecilia S.A. Ecuador.
2. Centro de Investigación Biomédica (CENBIO). Universidad. UTE. Ecuador.
3. Hospital de Especialidades Carlos Andrade Marín.
Ecuador.
4. Hospital General del Sur de Quito. Ecuador.
Doi: https://doi.org/10.23936/pfr.v5i3.181
Recibido: 05/10/2020 Aprobado: 23/11/2020
Resumen
La Lipodistrofia Congénita Generalizada o síndrome de Berardinelli – Seip, es un trastorno autosómico recesivo, con baja prevalencia a nivel mundial. Se caracteriza por lipoatrofia, características acromegaloides, hepatomegalia, hipertrigliceridemia, insulinorresistencia (criterios principales); miocardiopatía hipertrófica, deterioro intelectual, hirsutismo, pubertad precoz, quistes óseos, flebomegalia (criterios menores). Ésta patología se clasifica en 4 tipos, de acuerdo al gen alterado y por algunos síntomas característicos adicionales; tipo 1 (gen AGPAT2); tipo 2 (gen BSCL2); tipo 3 (gen CAV1); y tipo 4 (gen PTRF). Se realiza el abordaje de una adolescente que clínicamente cumple criterios clínicos diagnósticos, es necesario la verificación diagnóstica genética.
Palabras clave: Lipodistrofia Generalizada Congénita, Resistencia a la Insulina, Hipertrigliceridemia.
Generalized Congenital Lipodystrophy. Case report
Abstract
Congenital Generalized Lipodystrophy or Berardinelli-Seip syndrome, is an autosomal recessive disorder, with low prevalence worldwide. It is characterized by lipoatrophy, acromegaloid characteristics, hepatomegaly, hypertriglyceridemia, insulin resistance (main criteria); hypertrophic cardiomyopathy, intellectual deterioration, hirsutism, precocious puberty, bone cysts, phlebomegaly (minor criteria). This pathology is classified into 4 types, according to the altered gene and by some additional characteristic symptoms; type 1 (AGPAT2 gene); type 2 (BSCL2 gene); type 3 (CAV1 gene); and type 4 (PTRF gene). The approach of an adolescent who clinically meets clinical diagnostic criteria is performed, genetic diagnostic verification is necessary.
Keywords: Lipodystrophy, Congenital Generalized; Insulin Resistance; Hypertriglyceridemia
Introducción
La Lipodistrofia Congénita Generalizada (LCG) o Síndrome de Berardinelli-Seip es un trastorno genético de carácter autosómico recesivo muy raro que presenta una prevalencia mundial de 1 en 10 millones de habitantes (1). En Europa su prevalencia está estimada en 1/400.000 habitantes mientras que en Estados Unidos se ha registrado 1/12'000.000. Datos estadísticos a nivel de Latinoamérica es muy limitado, en el Ecuador no existen reportes sobre esta patología (2–4).
Está caracterizado principalmente por lipoatrofia, características acromegaloides, hepatomegalia, hipertrigliceridemia, insulinorresistencia (criterios principales); miocardiopatía hipertrófica, deterioro intelectual IQ 50-70/IQ 35-50, hirsutismo, pubertad precoz, quistes óseos y flebomegalia (criterios menores) (4,5). El diagnóstico se establece en un probando con tres criterios principales, o dos criterios principales más dos o más criterios menores (5).
Esta patología se clasifica en 4 tipos, de acuerdo al gen alterado y por algunos síntomas característicos adicionales: LCG tipo 1, se debe a mutaciones en el gen AGPAT2 (9q43), codifica la enzima (1-acilglicerol-3-fosfato-O-aciltransferasa-2) encargada de la síntesis de triglicéridos, es prevalente en individuos afroamericanos, con una presentación clínica leve entre la segunda o tercera década de la vida (6). LCG tipo 2, se debe a mutaciones en el gen BSCL2 (11q13), codifica la proteína seipina implicada en adipogénesis, es prevalente en ciertas áreas de Europa y Oriente Medio, tiene una expresión fenotípica más grave con una presentación desde el nacimiento o primera infancia, mayor prevalencia de discapacidad intelectual y miocardiopatía, además de muerte prematura (7).
LCG tipo 3, se debe a mutaciones homocigóticas en el gen CAV1 (7q31), que produce caveolina una proteína importante para la diferenciación de adipocitos, clínicamente mantiene características similares a LCG1 y LCG2 con un grado de lipodistrofia intermedio entre los dos fenotipos (8). LCG tipo 4, se debe a mutaciones en el gen PTRF (17q21) codifica a cavin tipo 1, proteína necesaria para la biogénesis de caveolas enroladas en el transporte transmembrana del adipocito, el cuadro clínico incluye lipodistrofia moderada, miopatía congénita, disfunción esofágica, estenosis pilórica, inestabilidad atlantoaxial, prolongación del intervalo QT con taquicardia ventricular inducida por el ejercicio y muerte súbita (9).
En Ecuador no se ha reportado casos con esta patología, tampoco existen laboratorios con la capacidad resolutiva necesaria para realizar estudios genéticos dirigidos a la conformación de la hipótesis diagnóstica de acuerdo a las características clínicas obtenidas.
La descripción del presente caso es abordado de acuerdo a los hallazgos clínicos encontrados en una adolescente de 15 años, sugestivos de una Lipodistrofia Congénita Generalizada tipo 2 debido a su peculiaridad. El principal objetivo de este informe de caso es el registro del mismo, la correlación del cuadro clínico con la evidencia de la literatura encontrada y la consideración de ciertas enfermedades raras o huérfanas por parte del estado con la finalidad de brindar un diagnóstico confirmatorio (11) y tratamiento adecuado.
CASO CLÍNICO
Se presenta el caso clínico de una adolescente de 15 años que inicialmente presentó poliuria, polidipsia y polifagia de 6 meses de evolución asociados a pérdida de peso en un mes, con niveles de glucemia en sangre periférica de 500 mg/dl. Como antecedentes personales, hija de un matrimonio sin grado de consanguinidad aparentemente, hija de madre soltera, sus tres hermanos dependientes de otro compromiso fallecieron debido a trastornos de la coagulación sin causa demostrada.
Producto de un embarazo sin complicaciones y sin controles, nacimiento pretérmino en parto intradomiciliario con bajo peso al nacimiento e hiperbilirrubinemia, por lo que recibió cuidados iniciales en casa asistencial con fototerapia e incubación, mal desarrollo pondoestatural, desarrollo psicomotor normal, al momento con vacunas actualizadas para su edad.
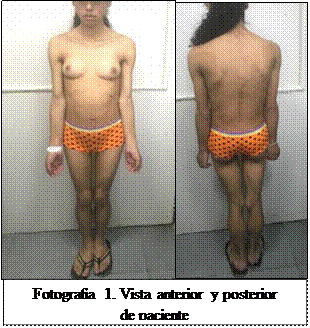
Entre los antecedentes patológicos se encontraron epistaxis recurrentes, procesos respiratorios a repetición, incontinencia urinaria al esfuerzo, laparotomía exploratoria por apendicitis complicada con peritonitis a los 9 años y amenorrea primaria como antecedentes ginecológicos. Su desarrollo mental aparentemente normal, sin embargo refirió haber perdido interés en estudios. Antecedentes familiares de abuelo materno con insuficiencia renal grado III por hipertensión arterial no controlada adecuadamente y abuela materna con bocio el cual fue extirpado quirúrgicamente.
En el examen físico, sus signos vitales normales, peso de 46,1 kg (percentil 19), talla 158 cm (percentil 26) e IMC 18,5 kg/m2 (percentil 27). Paciente impresionaba apariencia pseudoatlética junto con facies sindrómica con características acromegaloides con un rostro triangular, arcos superciliares muy marcados, mentón en punta, escápulas aladas, ausencia de grasa subcutánea en extremidades, cara y tronco. Neurológicamente se mantuvo con funciones mentales superiores y praxias conservadas. La valoración oftalmológica reveló astigmatismo sin evidencia de retinopatía.
En cuello y axilas distribución de acantosis nigricans, evaluación cardiovascular fue normal. Glándulas mamarias Tanner III. Abdomen con presencia de cicatriz queloide vertical infraumbilical sin evidencia de visceromegalias y genitales femeninos Tanner IV con presencia de clitoromegalia. Al examen musculoesquelético se observó marcha disbásica con limitación a la abducción de caderas, flebomegalia, musculatura bien definida y aumento de tono muscular generalizado.
Se valoró el coeficiente intelectual (CI) con resultado de 50 (limítrofe) mediante el test de WISC-IV (Escala de inteligencia de Wechsler para niños – IV).
En exámenes paraclínicos laboratoriales previos se encontró glucosa en sangre periférica en ayunas de 200 mg/dL, glucosa postprandial en sangre periférica de 250 mg/dL, análisis de orina con glucosuria (+) sin cetonuria. La monitorización frecuente de la glucosa en sangre reveló hiperglucemia persistente con valores entre 180-300 mg/dL. Insulina sérica de 58.3 μIU / ml (< 24,9 μUI/ ml), péptido C 8.6 ng/mL (1.1 – 5 ng/mL) lo cual sugiere resistencia a la insulina. El nivel de hemoglobina glicosilada (HbA1c) fue 12,95% (4,3-5,8%).
Su perfil lipídico mostró hiperlipidemia mixta con colesterol 295 mg/dL (135 - 200 mg/dL), lipoproteína de alta densidad (HDL) 35 mg/dL (135 - 200 mg/dL), lipoproteína de baja densidad (LDL) 106,4 mg/dL (135 - 200 mg/dL), triglicéridos 875 mg/dL (44-135 mg/dL). La biometría hemática e índices hematológicos fueron normales junto con electrolitos (Cl, Na, K, P, Ca total e iónico, Mg). Tiempos de coagulación normales, tiempo de protrombina (TP) 11.5’’ (11.0" - 14.5"), tiempo de tromboplastina (TTP) 30,0'' (20 - 33.3'')
La química sanguínea reveló niveles de úrea de 25,5 mg/dL (10 - 50 mg/dL), creatinina 0.69mg/dL (0.5 - 1.4 mg/dL), ácido úrico 9 mg/dL (3.4 - 7 mg/dL), amonio 82 ug/dL (18.7-86.9 ug/dL) . En el perfil hepático el aspartato amino transferasa (AST) 136.98 U/L(8-33 U/L), alanino aminotransferasa (ALT) 111 U/L (7-55 U/L), proteína C reactiva (PCR) 1,19 mg/dL (0 - 0.8 mg/dL), procalcitonina (PCT) 0.22 ng/ml (0 - 0.05 ng/ml), lactato deshidrogenasa (LDH) 369 UI/l (240 – 480 UI/l), proteínas totales 8.90 g/dL, (6.6 - 8.7 g/dL), albúmina 5,6 g/dL, ( 3.2 - 4.5 g/dL). Gasometría arterial con acidosis metabólica.
Perfil Endocrinológico: Cortisol AM 14,46 (mcg/dL), progesterona 0,13 ng/m, hormona folículo estimulante (FSH) 6.72 mUI/ml, hormona luteinizante (LH) 4.38 mUI/ml, 17-beta estradiol 22,25 pg/ml, factor de crecimiento insulínico tipo 1 (IGF-1) 243 ng/ml (111 - 996 ng/ml). Perfil tiroideo TSH 1.52 uUI/mL (0.48 - 4.17 uUI/mL), T4 libre 0.49 ng/dL (0.83 - 1.43 ng/dL),T3 libre 3.08 pmol/L (2-7pmol/L), anti TPO negativo, tiroglobulina normal 15 ng/ml (< 50 ng/ml).
Elemental microscópico de orina (EMO) no infeccioso, GRAM negativo, presencia de proteínas en orina 719 mg/L (< 150 mg/L), microalbuminuria cuantitativa 500.32 mg/L (30 mg/L-300 mg/L), índice albúmina Creatinina 483.4 mg/g (28-217 mg/dl), creatinina en orina 103,5 mg. Tasa de filtración glomerular preservada (98 ml/min), cociente proteinuria / creatinuria 0,6 significativa.
Estudios complementarios, electrocardiograma sin datos de dilatación o hipertrofia de cavidades. Ecocardiograma con insuficiencia tricuspídea leve: TAPSE (desplazamiento sistólico del plano del anillo tricuspídeo) 17 mm, IT (Insuficiencia tricuspídea) 28, PSAP (Presión sistólica de la arteria pulmonar) 38 mmHg, paciente con corazón sano, grosor de paredes cardíacas dentro de parámetros normales y presión pulmonar limítrofes.
Ecografía abdominal normal sin presencia de daño hepático ni megalias, ecografía renal con inflamación de médula sin ectasias, presencia de quiste simple de 14 mm en riñón izquierdo, ecografía de tiroides reveló nódulo tiroideo en lóbulo derecho, según la clasificación ATA (American Thyroid Association) intermedia, ecografía ginecológica dentro de parámetros normales. Resonancia magnética cerebral sin alteraciones. Rx de manos con edad ósea de 16 años y edad cronológica de 15 años 5 meses.
El tratamiento fue multidisciplinario, farmacológico basado en fibratos, IECAs, biguanidas, insulinoterapia de corta, intermedia y larga duración, además de terapia de suelo pélvico más antimuscarínicos, y tratamiento no farmacológico con terapia nutricional y actividad física. La presentación del caso clínico realizado cumplió con los principios éticos de la Declaración de Helsinki (10).
DISCUSIÓN
La lipodistrofia congénita generalizada fue descrita por primera vez en Brasil por Berardinelli y en Noruega por Seip en 1959 (12). Es una patología de baja frecuencia a nivel mundial y de prevalencia desconocida en Ecuador. Clínicamente se caracteriza por deficiencia de grasa corporal, diabetes e hiperinsulinemia secundaria, resultante en incapacidad de los tejidos de responder a la insulina.
De acuerdo a las características clínicas del informe de caso clínico expuesto y según la literatura se ha determinado una lipodistrofia congénita generalizada por la presencia de cuatro criterios mayores como lipoatrofia, hipertrigliceridemia, insulinorresistencia y características acromegaloides y un criterio menor como flebomegalia (5). Manifestaciones como hepatomegalia, miocardiopatía hipertrófica, hirsutismo, pubertad precoz y quistes óseos no se evidenciaron en nuestra paciente. Las peculiaridades clínicas como el CI-50, edad de presentación en adolescencia, mal desarrollo pondoestatural en el nacimiento y primera infancia, se lo ha catalogado como Lipodistrofia Congénita Generalizada Tipo 2 (5).
Fisiopatológicamente las alteraciones genéticas primarias en la biogénesis de lípidos producen diabetes y dislipidemia. Los niveles de leptina y adiponectina se encuentran implicados en la sensibilidad a la insulina, control glucémico, dislipidemia y esteatosis hepática (13).
El manejo de la LCG se centra en controlar la diabetes, mejorar la resistencia a la insulina y reducir los niveles de triglicéridos. El tratamiento es dirigido al control de distintos problemas mas no del síndrome en sí, mismos que consisten en la restricción de la ingesta de grasas y en controlar la glucemia el cual se lo manejó en nuestro caso mediante control nutricional, actividad física, biguanidas, insulinoterapia y fibratos de acuerdo a la disponibilidad de fármacos en nuestro país. El tratamiento indicado en nuestra paciente mostró una mejoría en su condición clínica, sin embargo el control con leptina recombinante es necesario de acuerdo a las recomendaciones y evidencia en pacientes con la misma patología (5), del cual nuestro país no es tributario.
En nuestro caso índice no se encontró hallazgos significativos en el estudio ecográfico de hígado a pesar de que los niveles de triglicéridos y transaminasas hepáticas fueron elevados. La evidencia de glucosuria, proteinuria e índice de proteína/creatinina significativo en orina fueron indicativos de alteración en la función renal, complicación frecuente ante la presencia de niveles de glucemia elevados persistentes (14,15), por lo cual el manejo inicial de protección renal fue a base de inhibidores de la enzima convertidora de angiotensina con un régimen a bajas dosis. Finalmente, por los antecedentes de incontinencia urinaria fue necesario el seguimiento por urología para terapia con antimuscarínico y fisioterapia de piso pélvico.
Es muy importante el tratamiento multidisciplinario, para su adecuado seguimiento se dispone actualmente de insulina para administración subcutánea, que consideramos un punto importante a tratar por sus efectos secundarios luego de su administración por varias ocasiones, y que por su condición de lipoatrofia, la cantidad de administración de insulina no es metabolizada adecuadamente.
Adicionalmente podemos destacar que la semiología juega un rol fundamental para el abordaje de patologías endocrino-metabólicas que son prevalentes en nuestro medio, por tanto los profesionales sanitarios que tienen un primer encuentro con pacientes con este tipo de problemas deberán hacer una investigación minuciosa a través de la anamnesis conjuntamente con un examen físico detallado para encontrar la etiología de este tipo de enfermedades que en el caso previamente descrito corresponde a una causa de tipo genético.
La financiación limitada por parte del estado dirigido a investigación para la realización de estudios genéticos para trastornos de esta índole son un limitante. La atención a todos los pacientes debe ser eficiente y eficaz con la finalidad de evitar complicaciones o agravamiento de su cuadro clínico, tratando de brindar una mejor calidad de vida. Sin duda el reporte de casos como este para que de una u otra manera sea de lectura crítica por los profesionales de la salud y diferentes autoridades de la entidad estatal y sanitaria, tratando de ser un nexo para ayuda a la población con este tipo de patologías.
Al ser una condición rara existe evidencia limitada, por lo cual es prioritario mayor investigación por parte de expertos así como el registro del mismo.
CONCLUSIONES
El reporte de este caso, dado su cuadro clínico fueron sugestivos de una Lipodistrofia Congénita Generalizada, por lo cual el personal sanitario debe estar previamente capacitado para el diagnóstico clínico oportuno y un consecuente manejo multidisciplinario para este tipo de condiciones y así evitar complicaciones, a pesar de que al momento no exista disponibilidad de estudios genéticos específicos de esta índole en el país.
La participación por parte de autoridades del estado es primordial, encaminado a brindar una mejor calidad de vida, de forma especial a la población de bajos recursos económicos que presentan ciertas enfermedades raras como la LCG y necesitan de un tratamiento especial dirigido a la enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1. Garg A. Medical progress: acquired and inherited lipodystrophies. N Engl J Med. 2004; 350 (12): 1220-34.
2. Orphanet [Internet]. Van, L; [Actualizado Enero 2009; Citado 14 abr 2020]. Lipodistrofia congénita de Berardinelli-Seip [Aprox 4 pantallas]. Disponible en: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=528.
3. Informes Periódicos de Orphanet -Prevalencia de las enfermedades raras: Datos bibliográficos -Enero2020-Número 2. Disponible en: https://www.orpha.net/orphacom/cahiers/docs/ES/Prevalencia_de_las_enfermedades_raras_por_prevalencia_decreciente_o_casos.pdf. 4. Van Maldergem, L., Berardinelli-Seip congenital lipodystrophy. Orphanet encyclopedia. November 2001. http://www.orpha.net/data/patho/GB/uk-berard.pdf.
5. Van Maldergem L. Berardinelli-Seip Congenital Lipodystrophy. 2003 Sep 8 [Updated 2016 Dec 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
6. Garg, A. Et, al. A Gene for Congenital Generalized Lipodytrophy Maps to Human Chromosome 9q34. The Journal of Clinical Endocrinology & Metabolism, Volume 84, Issue 9, 1 September 1999, Pages 3390–3394.
7. Magré, J., Delépine, M., Khallouf, E. et al. Identification of the gene altered in Berardinelli–Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001; 28, 365–370.
8. Kim CA, Delépine M, Boutet E, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129–1134.
9. Hayashi YK, Matsuda C, Ogawa M, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119(9):2623–2633.
10. Asociación Médica Mundial. Declaración de Helsinki de la Asociación Médica Mundial. Principios éticos para las investigaciones médicas en seres humanos [sede web]*. Brasil: 64a Asamblea de la Asociación Médica Mundial; 9 de julio del 2018 [acceso 17 de diciembre del 2019]. Disponible en: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-formedical-research-involving-human-subjects/.
11. Jéru, I., Vatier, C., Araujo-Vilar, D., Vigouroux, C., and Lascol, O. Clinical Utility Gene Card for: Congenital Generalized Lipodystrophy. European Journal of Human Genetics. 2016; 24
12. Metwalley KA, Farghaly HS. Berardinelli-Seip syndrome type 1 in an Egyptian child. Indian J Hum Genet. 2014;20(1):75–78. doi:10.4103/0971-6866.132762
13. Bhayana S, and Hegele RA. La base molecular de las lipodistrofias genéticas. Clin Biochem. Mayo de 2002; 35 (3): 171-7.
14. Cardona-Hernández R., Suárez-Ortega L., Torres, M. Lipodistrofia congénita generalizada versus síndrome de Berardinelli-Seip. An Pediatr (Barc). 2011;74:423-410
15. Daher, E. Et, al. Berardinelli syndrome. A case report with fatal outcome. Invest Clin 49(2): 251 - 255, 2008


